(1) Molecular Genetics Laboratory, Department of Psychiatry, Kiel University Hospital, Kiel, Germany; (2) Department of Psychiatry, National University Hospital, Reykjavik, Iceland; (3) Department of Psychiatry, University of Minnesota, Minneapolis, USA
Corresponding author: Dr. Hans W. Moises, Molecular Genetics Laboratory, Department of Psychiatry, University of Kiel, Niemannsweg 147, 24105 Kiel, Germany; Phone: +49(431) 597-2681, Fax: +49(431) 597-2572, Email: moises@psychiatry.uni-kiel.de
Numerous studies support the genetic and the neurodevelopmental hypotheses of schizophrenia. However, association with a neurodevelopmentally important gene has not yet been demonstrated in schizophrenia. To follow up a lod score of 2.43 at 8p21-22 obtained during a genome scan in schizophrenia families from Iceland, we searched for allelic association in the region. Three-hundred and twelve individuals from Iceland (151 unrelated schizophrenic patients and 161 family members as control) participated in the study. The diagnoses were made independently by two experienced psychiatrists according to DSMIIIR criteria. The genotyping data of markers at 8p were analysed by a chi-square test. Highly significant evidence for allelic association was obtained for two markers, D8S1777 and D8S1719, which flank the neuregulin 1 gene (p = 6.6E-7 and 8.0E-6, respectively). D8S1777 showed the strongest association with schizophrenia (X = 36.00). The closest gene to D8S1777 is neuregulin 1 within a distance of 1.03 Mb. Neuregulin plays an important role in neurodevelopment during the second trimester, a prenatal period which shows evidence for neurodevelopmental disturbances in schizophrenia. In the adult brain, neuregulin is involved in synaptic plasticity and long-term potentiation, mechanisms relevant for memory, which is impaired in schizophrenia. In conclusion, position as well as function strongly supports the neuregulin-1 gene (NRG1) as a susceptibility gene for schizophrenia. The final determination, however, depends on the detection of functional polymorphisms of NRG1 associated with schizophrenia. If borne out by further research, activation of the neuregulin-ErbB network could be used as a basis for a new form of rational treatment for schizophrenia.
The
current understanding of the etiology of schizophrenia is based primarily on the genetic and the neurodevelopmental
hypotheses [1]
. The
neurodevelopmental hypothesis of schizophrenia was first proposed in the 20th
century by Kraepelin, Southard and others as
explanation for neuropathological abnormalities found in the brains of some
schizophrenic patients [2]
.
The hypothesis was revived in 1982 by Feinberg who
suggested a faulty programmed synaptic elimination during adolescence as cause
of schizophrenia [3]
, in 1987
by Murray and Lewis [4]
and in 1995 by Weinberger [5]
to account for more recent research findings. The strength of the
neurodevelopmental hypothesis is its ability to explain a variety of results
obtained in clinical, epidemiological, neuropathological, and imaging studies,
e.g. minor
physical anomalies [6]
, maternal
virus infections [7,
8]
, winter
births [9,
10]
,
obstetric complications [11,
12]
, low birth weight [13,
14]
, mis-sized or disorganised neurons [15]
, and volume reductions
of grey matter [16,
17]
(for review, [18]
).
The genetic hypothesis is supported by numerous family, twin, adoption, and linkage studies [19, 20] which are all in agreement with the multigenic model of schizophrenia elaborated in 1967 by Gottesman & Shields [21] . In the search for susceptibility genes for schizophrenia, genome scans and allelic association studies are currently employed. The first linkage study in schizophrenia was reported in 1958 by Constantinidis [22] , and the first replicated linkage finding was in 1979 by Turner using HLA markers (lod 2.57) [23] . The first completed genome scans were accomplished in 1994 by Byerley using an RFLP map [24] and in 1995 by Moises and Helgason using Généthon´s modern genetic map of informative markers in Icelandic families [25] . In 1994 we obtained evidence for linkage of schizophrenia to D8S269 by two-point lod score analysis (lod 2.43) [26] although a non-parametric linkage test (WRPC) gave only a weak signal (p = 0.05) [25] . Evidence for linkage of schizophrenia to 8p21-22 was obtained by several groups of investigators [25-35]. Following-up our 1994 and 1995 linkage finding at 8p [25, 26] , we started in 1996 to perform fine mapping studies of the region and to employ a twosome approach advocated by John H. Edwards from Oxford in an Icelandic sample. In 1998 we obtained significant evidence for allelic association between schizophrenia and D8S1777 (p = 0.00004 after Bonferroni correction). In 1999 we presented this significant finding at The VIIth World Congress of Psychiatric Genetics in Monterey (California) without releasing the name of the significant marker [36] . To identify the gene responsible for the significant association, we genotyped more markers in the neighbourhood of D8S1777. The improvement of the map in the region by the Human Genome Project revealed a close proximity between D8S1777 and neuregulin 1, a neurodevelopmental gene. Here we present the results of our fine mapping study showing a connection between the genetic and the neurodevelopmental hypotheses of schizophrenia.
The
investigation was approved by the local Ethic Committee. All the participants
gave informed consent.
A
total of 312 individuals were investigated from the Icelandic population
consisting of 151 unrelated schizophrenic patients (103 males, 48 females) and
161 of their family members (129 mothers, 13 fathers, 10 sons and 9 daughters)
as internal control. Using twosomes instead of trios is a simplified mapping
approach for common disorders advocated by John H. Edwards from Oxford
University. Affection status was defined by the diagnosis of schizophrenia (N =
155) or schizoaffective psychosis (N = 1). Life-time diagnoses were given
independently by two experienced psychiatrists according to DSM-IIIR criteria [37]
using information from medical records of the Department of Psychiatry
at the University Hospital in Reykjavik in Iceland. Four of the controls (parents)
were affected by schizophrenia. The age of patients ranged from 18 to 85 years,
average age was (39.2) years, while the age of the parents ranged from 47 to 85
years, average age was 67.5 years. The age of the children ranged from 17 to 51
years, average age 32.7 years. The age at onset of disease ranged from 10 to 50
years, the average age at onset was 20.7 years.
Genomic
DNA was extracted from blood using the Pure Gene DNA extraction kit from Gentra
Systems Inc, Minneapolis, MN, USA. Eleven markers were genotyped in the
neigbourhood of D8S1777: D8S1734, D8S1786, D8S1725, D8S1770, D8S1839, D8S1820, D8S1777,
D8S1719, D8S1121, D8S1722, and D8S255. The markers and primer
sequences are described in the Genome Database (GDB).
Polymerase chain reaction (PCR) was performed in a 20 ml
total reaction volume containing 40 ng of
genomic DNA, 5 mM
of each primer (the forward primer was fluorescently labelled with the dye FAM
for all 3 markers) , 2.5 mM of dNTP’s and 1.5 mM MgCl2 using 96 well
polycarbonate microtitre plates (Techne Inc, NJ, USA).The amplification was done
using the ‘Hot Start’ procedure which consists of a denaturation step at 97°
C for 5 min followed by addition of 0.5 units of Taq polymerase (Renner GmBH,
Darmstadt, Germany), followed by 32 cycles of denaturation at 94° C for 40 sec,
annealing at 55° C for 40 sec and extension at 72° C for 40 sec and a final
extension for 5 mins at 72 °C. Pooled PCR products from the markers were
electrophoresed on 6 % denaturing acrylamide gels using an automated DNA
sequencer (ABI, 377 Perkin Elmer, Applied Biosystems, Forster City, CA, USA).
Fragment sizing was performed using the GENESCAN Software version 2.0.2 (Perkin
Elmer Applied Biosystems). Genotyping was done blindly in regard to the
affection status.
Searching
for allelic association between schizophrenia and marker alleles, we performed a
c2 test in doublettes. The use of doublettes is a
mapping strategy for common disorders advocated by John H. Edwards of Oxford
University, termed PQR or twosome approach. It is based on the doublette
approach developed by Bernstein in 1931 which was later extended to sib-pairs by
Penrose [38]
. The use of twosomes has the advantage of saving resources better
directed to increasing the sample size. Twosomes consist of affecteds and one of
their parents or children as control. In the PQR approach, the frequency of the
allele not transmitted from the first parent to the affected (P) serves as
family-based control and is compared to the frequency of the transmitted allele
from the second parent (R). The allele transmitted to the affected from the
first parent is designated as Q. Likewise in affected-child pairs, Q is
identical in both individuals, R the non-identical allele of the affected, and P
the control allele in the child. A similar approach has been proposed for trios
by Falk and Rubinstein to easily obtain a reliable control sample by using as
control both parental alleles not present in the affected (Hapoltype Relative
Risk, HRR approach) [39]
. However, a simple c2
test cannot be employed in the HRR approach or the widely used Transmission
Disequilibrium Test (TDT) since transmission and non-transmission are not
independent. Every increase in transmission produces a decrease in
non-transmission count. In the PQR method, by contrast, transmission and
non-transmission occur independently in two different parents permitting the use
of simple and additive c2
tests comparable to a case-control design. A priori the twosome method compares
favorably to other study designs. It seems to be an economic, powerful, easy and
reliable strategy.
For
statistical analysis of individual markers, twosomes were discarded when it was
impossible to deduce which allele has been transmitted, e.g. the affected was
homozygous or the affected and control subject had identical genotypes or
genotypes were missing. For individual markers, allele counts were tabulated in
contingency tables consisting of two rows (P & R) and columns representing
the alleles. Alleles with allele counts £
5 were removed. The c2 value
associated with the contingency table was calculated in the usual way as the sum
over all cells of the squared difference between observed and expected value
divided by the expected value. The expected values were calculated conditional
on the row and column totals. The expected value for a cell was the total for
its row multiplied by the total for its column divided by the overall total
number of observations. Sham and Curtis’s program CLUMP version 1.9 was
employed for calculating the c2 value
(designated as T1 statistic) [40]
. The program uses Monte Carlo (MC) simulations to assess the number of
times a simulated 2-by-m table with the same row and column total as the
contingency table yields a c2
value equal or larger than the one obtained from the real table. The MC simulation has the advantage
that a reliable assessment of the significance is given even if small expected
values in some cells do not follow the expected distribution of the c2
statistic and that no special account has to be taken
of continuity corrections. Hundred million MC simulations were used to estimate
the p
value for
D8S1777 and D8S1719.
The
relative incidence (X or relative risk, RR) of alleles with a count of more than
5 was determined according to Woolf’s formula [41]
. Jurg Ott’s program RelRisk version 2.33 was employed for calculation.
For individual markers, the maximum value of X was used as a measure for the
strength of the allelic association. Allelic association between pairs of
markers was tested by employing Abecasis and Cookon’s program GOLD version 1.0
[42]
which uses as input founder haplotypes estimated by Sobel and Lange’s
program Simwalk2 version 2.82 [43]
. Distances between markers and genes were obtained from The Unified
Database (UDB) [44]
which is based on the human genome sequence of the National Center for
Biotechnology Information (NCBI).
The
anomyzed genotyping data are given in the appendix to
enable other workers in the field to test the power of different statistical
methods or to obtain more power for their analyses by simply adding our data to
theirs.
Table
1 displays the allele counts for individual markers, the maximum value of X (=
RR), the results of the chi-square
(c2)
analysis,
and p values derived from Monte Carlo simulations.
Table 1. Results of chi-square (c2) analysis for allelic association between schizophrenia and markers on 8p.
|
Alleles |
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
19 |
20 |
X |
c2 |
P# |
|
D8S1734 |
P |
2 |
7 |
11 |
10 |
11 |
5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1.54 |
2.70 |
0.63 |
|
|
R |
2 |
8 |
6 |
8 |
15 |
7 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
D8S1786 |
P |
1 |
14 |
12 |
23 |
6 |
4 |
12 |
6 |
13 |
1 |
12 |
0 |
|
|
|
|
|
|
|
|
1.30 |
6.61 |
0.58 |
|
|
R |
0 |
17 |
17 |
18 |
7 |
6 |
16 |
2 |
7 |
0 |
12 |
2 |
|
|
|
|
|
|
|
|
|
|
|
|
D8S1725 |
P |
1 |
7 |
18 |
32 |
18 |
1 |
1 |
2 |
0 |
|
|
|
|
|
|
|
|
|
|
|
2.81 |
16.92 |
0.001 |
|
|
R |
0 |
17 |
25 |
11 |
26 |
0 |
0 |
0 |
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
D8S1770 |
P |
11 |
0 |
3 |
25 |
3 |
0 |
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
2.59 |
6.82 |
0.04 |
|
|
R |
19 |
0 |
2 |
14 |
7 |
1 |
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
D8S1839 |
P |
0 |
1 |
2 |
8 |
26 |
7 |
4 |
1 |
2 |
|
|
|
|
|
|
|
|
|
|
|
2.66 |
18.99 |
0.001 |
|
|
R |
1 |
0 |
5 |
17 |
6 |
7 |
8 |
3 |
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
D8S1820 |
P |
1 |
33 |
12 |
1 |
14 |
12 |
5 |
3 |
|
|
|
|
|
|
|
|
|
|
|
|
2.39 |
16.36 |
0.001 |
|
|
R |
3 |
14 |
6 |
1 |
26 |
17 |
11 |
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
D8S1777 |
P |
0 |
19 |
1 |
1 |
2 |
0 |
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
36.00 |
24.55 |
0.00000066 |
|
|
R |
1 |
3 |
0 |
15 |
1 |
1 |
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
D8S1719 |
P |
0 |
26 |
5 |
4 |
2 |
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5.34 |
21.84 |
0.000008 |
|
|
R |
5 |
5 |
17 |
8 |
3 |
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
D8S1121 |
P |
0 |
6 |
10 |
20 |
18 |
27 |
1 |
6 |
2 |
1 |
|
|
|
|
|
|
|
|
|
|
2.59 |
5.17 |
0.52 |
|
|
R |
2 |
13 |
9 |
18 |
17 |
20 |
0 |
7 |
5 |
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
D8S1722 |
P |
2 |
1 |
0 |
3 |
3 |
6 |
9 |
19 |
1 |
10 |
11 |
0 |
6 |
23 |
6 |
9 |
7 |
2 |
0 |
1 |
2.72 |
13.28 |
0.26 |
|
|
R |
0 |
0 |
1 |
6 |
6 |
5 |
13 |
13 |
0 |
10 |
12 |
1 |
15 |
25 |
1 |
7 |
3 |
0 |
1 |
0 |
|
|
|
|
D8S255 |
P |
5 |
6 |
3 |
16 |
20 |
2 |
4 |
4 |
1 |
|
|
|
|
|
|
|
|
|
|
|
2.79 |
7.37 |
0.30 |
|
|
R |
5 |
10 |
2 |
10 |
14 |
5 |
5 |
10 |
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Only
alleles with a frequency exceeding five were used for chi-square
analysis. # = without Bonferroni correction.
Table
2 shows the results of the analysis for allelic association between paired
markers with a p
value of less
or equal 0.01
as well as for D8S1777 with the other markers.
Table
2.
Results
of chi-square
(c2)
analysis
by GOLD for allelic association between paired markers.
|
Marker
A |
Marker
B |
N |
df |
|
p |
D' |
|
D8S1777 |
D8S1734 |
242 |
10 |
14.84 |
0.13 |
0.18 |
|
D8S1777 |
D8S1786 |
394 |
10 |
9.21 |
0.51 |
0.13 |
|
D8S1777 |
D8S1725 |
394 |
8 |
12.97 |
0.11 |
0.07 |
|
D8S1777 |
D8S1770 |
398 |
4 |
0.75 |
0.94 |
0.02 |
|
D8S1777 |
D8S1839 |
317 |
8 |
9.17 |
0.32 |
0.13 |
|
D8S1777 |
D8S1820 |
386 |
8 |
9.19 |
0.32 |
0.12 |
|
D8S1777 |
D8S1719 |
352 |
6 |
8.74 |
0.18 |
0.37 |
|
D8S1777 |
D8S1121 |
380 |
10 |
9.78 |
0.45 |
0.13 |
|
D8S1777 |
D8S1722 |
392 |
14 |
26.98 |
0.02 |
0.17 |
|
D8S1777 |
D8S255 |
334 |
10 |
6.77 |
0.74 |
0.09 |
|
D8S1820 |
D8S255 |
350 |
20 |
39.47 |
0.006 |
0.14 |
|
D8S1719 |
D8S1121 |
375 |
12 |
59.72 |
0.0000 |
0.23 |
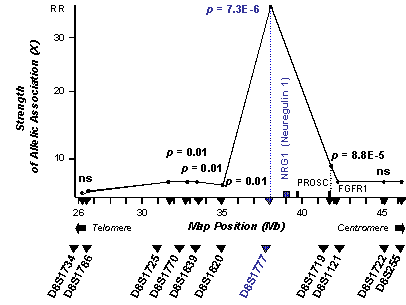
Figure 1 depicts the evidence for allelic association between schizophrenia and
the two markers flanking the NRG1 gene on chromosome 8p21-p22. Map distances are
derived from the UDB.
Figure 1. Evidence for allelic association of schizophrenia with markers on chromosome 8p. Map positions of markers and genes are obtained from the UDB and given in Megabases (Mb). The region between the highly significant markers D8S1777 and D8S1719 contains three HUGO approved genes according to the UDB: NRG1 (neuregulin 1), PROSC (proline synthetase co-transcribed – bacterial homolog), FGFR1 (fibroblast growth factor receptor 1) as well as hypothetical proteins and open reading frames (not shown). P values were obtained by Monte Carlo simulations and are Bonferroni corrected for multiple testing.
In
searching for schizophrenia susceptibility genes, we started in 1996 to apply
the twosome or PQR approach as an economic and efficient tool for the mapping of
the genes. Markers in the linkage region on chromosome 8p were genotyped in a
schizophrenia sample of twosomes from Iceland. In 1998, the chi-square
analysis of the genotyping data revealed a highly significant allelic
association of schizophrenia with D8S1777 (p
= 0.00004 after Bonferroni correction). In 1999 we reported the finding at the
VIIth World Congress of Psychiatric Genetics in Monterey (California) without
releasing the name of the significant marker [36]
. At that time, map position of markers and genes in the area kept
changing, the
genomic sequence of the region was not available,
and our attempt failed to organize a large-scale sequencing project of
the D8S1777 region ahead of the schedule of the Human Genome Project (HGP).
Waiting for the HGP to provide us with the relevant gene in the region we
genotyped more markers and more individuals to improve the fine mapping of the
susceptibility gene. The evidence for allelic association of schizophrenia with
D8S1777 continued to improve (p = 6.6E-7, see Table 1). Furthermore, we
obtained a significant result for another marker in the region, D8S1719 (p
= 8.0E-6, see Table 1). The latter is located 3,862,083
basepairs centromeric of D8S1777 according to the UDB map (see Figure
1).
Although both markers, D8S1777 and D8S1719, showed evidence for allelic
association among each other (D’ = 0.37), the chi-square
test was not significant (p = 0.18, see Table 2). Therefore, haplotype
analysis was not applied.
In
positional cloning, disease genes are identified by position, function and
finally mutation. In regard to position, our results place a susceptibility gene
for schizophrenia in the region between D8S1777 and D8S1719. These two
significant markers are 3.9
Mb apart according to the UDB (see Figure 1). This is an unusually large
distance for obtaining allelic association (AA) and nearly excludes the
susceptibility gene from the region telomeric of D8S1777 since a more telomeric
position would further increase the distance for AA between D8S1719 and
schizophrenia beyond 3.9 Mb. In addition, the next telomeric marker, D8S1820,
does not give evidence for AA although it is less than 3.9 Mb away from D8S1777
(3.0 Mb), the marker with the strongest AA (see Figure 1). Hence our results
place a susceptibility gene for schizophrenia within the region flanked by
D8S1777 and D8S1719. Within the region, the susceptibility gene must be closer
to the former than to the latter since the strength of association as measured
by X is with a relative risk (RR or X) of 36.00, more than 6.5 times higher at
D8S1777 than at D8S1719 with a RR of only 5.34 (see Table 1). The closest gene
to D8S1777 is the neuregulin-1 gene (NRG1) within a distance of 1.03 Mb (see Figure 1). The two
other HUGO approved genes in the region flanked by the significant markers are
PROSC (proline synthetase co-transcribed – bacterial homolog) and FGFR1 (fibroblast
growth factor receptor 1), all more distant from D8S1777 and therefore more
unlikely to be responsible for the AA with schizophrenia. Therefore, the position
suggests that NRG1 is the gene responsible for the strong allelic association
with schizophrenia obtained in our study.
Function
also strongly supports the neuregulin-1 gene as a susceptibility gene for
schizophrenia. Neuregulin is known to play an important role in neurodevelopment
during midgestation (for review, [45,
46]
). It is able to explain the evidence for neurodevelopmental disturbances
observed in schizophrenia (for review, [18]
[1,
47]
) which also seem to occur during midgestation [48]
.
Neuregulin
1 (NRG1) is expressed in neurons, glia, heart, liver, stomach, lung, kidney,
spleen and skin [46]
. Disruption of the NRG1 gene in mice results in aberrant branching
patterns of peripheral neurons, initial synapse formation followed by withdrawal
and degeneration of sensory and motor nerves [49]
, a process reminiscent of the neurodegenerational process observed in
schizophrenia [16,
50, 51]
. In schizophrenic patients degeneration of the second motor neuron
leading to an increase in muscle-derived serum creatine phosphokinase has been
reported by Meltzer and others [52-57]
. In the adult brain, neuregulin signaling is involved in the modulation
of synaptic plasticity and hippocampal long-term potentiation [58]
, brain mechanisms thought to be important for memory formation [59,
60]
. Memory impairment has not only been reported in the majority of
schizophrenic patients [61-64]
(for review [47]
) but also seems to be responsible for the characteristic symptomatology
of schizophrenia [65,
66]
. Furthermore, the fact that the receptors of neuregulin (erbB2, erbB3,
and erbB4) are
implicated in the etiology and progression of the majority of human carcinomas [67,
68]
could explain the observed negative association between schizophrenia
and cancer [69,
70]
. Moreover, neuregulin, also known as glial growth factor (GGF) [45]
is important for the development and survival of oligodendrocytes [46]
. A dysfunction of neuregulin could explain the evidence for a functional
deficiency of oligodendrocytes in schizophrenia obtained by Fienberg and
co-workers [71]
by using modern DNA microarray technology for a genome-wide gene
expression analysis in post-mortem brain tissue of schizophrenic patients.
Summing
up, position and function strongly support the neuregulin-1 gene (NRG1) as a
susceptibility gene for schizophrenia. The final determination, however, depends
on the detection of functional polymorphisms of NRG1 which are associated with
the presence of schizophrenia.
Alternative
splicing of NRG1 gives rise to multiple isoforms known as Neu differentiation
factor (NDF/heregulin), glial growth factor (GGF), and acetylcholine receptor
inducing activity (ARIA). Since neuregulins are EGF-like polypeptide growth
factors involved in neurodevelopment and cellular growth, our NRG1
finding is in full agreement with Weinberger’s neurodevelopmental hypothesis
[4,
5]
and Moises’ hypothesis of a cerebral protein-synthesis deficiency [47] in
schizophrenia. Briefly, the latter employed an evolutionary strategy
to identify susceptibility genes among the reported linkage regions of
schizophrenia. This approach identified among other genes neuregulin 1 (NRG1)
and several of the genes downstream in the signaling cascade of the
neuregulin-ErbB network [72]
such as insulin-like growth factor 1 (IGF1), phosphoinositide 3-kinase (PIK3),
and mitogen-activated protein kinases (MAPK). He concluded
that the common factor seems to be genetic and epigenetic variation of genes
involved in transcription, translation and signal transduction possibly leading
to a cerebral protein-synthesis deficiency [47]
. The hypothesis is in agreement with the fact that the neuregulin-ErbB
network represents a machinery developed for fine-tuning of signal transduction [72]
, that neuregulin-1 acts synergistically with the insulin-like growth
factor-1 (IGF1) [73]
, that the neuregulin effect is mediated by the phosphoinositide 3-kinase
(PIK3) [74]
, and finally that the binding
of neuregulin
to its receptors initiates a signaling cascade which culminates in the
activation of the mitogen-activated protein kinase (MAPK) pathway [75]
.
In
regard to therapy or prevention of schizophrenia, it is interesting to note that
brain neuregulin is increased by neuronal activity such as locomotion and
seizures [76]
and that an engineered neuregulin termed recombinant
human Glial Growth Factor 2 (rhGGF2) from Cambridge NeuroScience Inc. in
Cambridge Massachusetts (USA) is currently being developed by the Bayer Corporation for the
treatment of neurodegenerative
diseases such as multiple sclerosis and Parkinson’s disease. The results of
our investigation suggest, if borne out by further research, that activation of
the neuregulin-ErbB network by rhGGF2 or other means might be used in the future
to treat or prevent schizophrenia.
We
are indebted to Professor John H. Edwards, University of Oxford, for suggesting
the twosome approach. We are most grateful to the people of Iceland for their
support of an international research effort in schizophrenia. And we thank Dr. Paul
Ginsparg , the founder of the e-print archives at the Cornell University
Library, as well as the Stanford
University Library and the British Medical
Journal, the sponsors of the Netprint archives for Clinical Medicine &
Health Research, for providing the archives as electronic equivalent of
presenting the results at an international scientific conference with
the additional benefit that more scientists can encounter and comment on the
paper.
For physicists, this is a well-established routine.
In their view, “the way biologists are said to stake intellectual property
claims is intrinsically irrational --
that is, waiting for an official journal publication date, as though the work is
not intrinsically correct until officially "validated" ” (Ginsparg).
Since its inception in 1991, Paul Ginsparg’s e-print archives at the Los
Alamos National Laboratory have become a major forum for the dissemination of results in physics and
mathematics. By adopting their approach, we hope to encourage other researchers
in the life sciences to similarily follow the paradigm of the physical sciences
(see the preprint debate).
The
work was supported in part by the German Research Foundation (DFG), Bonn,
Germany.
AA;
allelic association
c2; chi-square
EGF;
epidermal growth factor
FGFR1;
fibroblast growth factor receptor 1
GGF;
glial growth factor
IGF1;
insulin-like growth factor 1
Lod;
lod score
HGP;
Human Genome Project
HUGO;
Human Genome Organisation
MAPK;
mitogen-activated
protein kinase
MC;
Monte Carol
Mb
= Megabases
NCBI; National Center for Biotechnological Information
NRG1;
neuregulin 1 gene
PIK3;
phosphoinositide 3-kinase
PROSC;
proline synthetase co-transcribed – bacterial homolog
rhGGF2
= recombinant human Glial Growth Factor 2
RR;
relative risk, relative incidence, X, a measure of the strength of association
TDT;
Transmission Disequilibrium Test
1. Moises, H.W.M., Zoega T., Li L., Hood L., Genes and neurodevelopment in schizophrenia, in Behavior Genetic Principles - Development, Personality, and Psychopathology: A Festschrift in Honor of Prof. Irving I. Gottesman, D.L. DiLalla, Editor. 2002, APA in press.
2. Southard, E.E., On the topographical distribution of cortex lesions and anomalies in dementia praecox, with some account of their functional significance. Am J Insanity, 1915. 71: p. 603-671.
3. Feinberg, I., Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J Psychiatr Res, 1982. 17: p. 319-334. [PubMed]
4. Murray, R.M. and S.W. Lewis, Is schizophrenia a neurodevelopmental disorder? Br Med J (Clin Res Ed), 1987. 295: p. 681-682. [PubMed]
5. Weinberger, D.R., From neuropathology to neurodevelopment. Lancet, 1995. 346(8974): p. 552-557. [PubMed]
6. McNeil, T.F. and E. Cantor-Graae, Minor physical anomalies and obstetric complications in schizophrenia. Aust N Z J Psychiatry, 2000. 34 (Suppl): p. S65-73. [PubMed]
7. Mednick, S.A., et al., Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psychiatry, 1988. 45:: p. 189-192. [PubMed]
8. Rantakallio, P., et al., Association between central nervous system infections during childhood and adult onset schizophrenia and other psychoses: a 28-year follow-up. Int J Epidemiol, 1997. 26: p. 837-
9. Machon, R.A., S.A. Mednick, and F. Schulsinger, Seasonality, birth complications and schizophrenia in a high risk sample. Br J Psychiatry, 1987. 151: p. 122-124. [PubMed]
10. McGrath, J.J. and J.L. Welham, Season of birth and schizophrenia: a systematic review and meta- analysis of data from the Southern Hemisphere. Schizophr Res, 1999. 35: p. 237-242. [PubMed]
11. Parnas, J., et al., Perinatal complications and clinical outcome within the schizophrenia spectrum. Br J Psychiatry, 1982. 140: p. 416-420. [PubMed]
12. McNeil, T.F., E. Cantor-Graae, and D.R. Weinberger, Relationship of obstetric complications and differences in size of brain structures in monozygotic twin pairs discordant for schizophrenia. Am J Psychiatry, 2000. 157: p. 203-212. [PubMed]
13. Kringlen, E., Schizophrenia in Male Monozygotic Twins. 1964, Oslo: Universitetsforlaget.
14. Wahlbeck, K., et al., Association of schizophrenia with low maternal body mass index, small size at birth, and thinness during childhood. Arch Gen Psychiatry, 2001. 58: p. 48-52. [PubMed]
15. Arnold, S.E., Cellular and molecular neuropathology of the parahippocampal region in schizophrenia. Ann N Y Acad Sci, 2000. 911: p. 275-292. [PubMed]
16. Henn, F.A. and D.F. Braus, Structural neuroimaging in schizophrenia. An integrative view of neuromorphology. Eur Arch Psychiatry Clin Neurosci, 1999. 249(Suppl 4): p. 48-56. [PubMed]
17. Shenton, M.E., et al., A review of MRI findings in schizophrenia. Schizophr Res, 2001. 49: p. 1-52. [PubMed]
18. Marenco, S., Weinberger, D.R., The neurodevelopmental hypothesis of schizophrenia: following a trail of evidence from cradle to grave. Dev Psychopathol, 2000. 12: p. 501-527. [PubMed]
19. Gottesman, I.I., Schizophrenia Genesis: The Origins of Madness. 1991, New York: W.H. Freedman.
20. Moises, H.W., Gottesman, I.I., Genetics, risk and personality factors, in Contemporary Psychiatry, F. Henn, Sartorius, H., Helmchen H., Lauter, N., Editor. 2000, Springer: Heidelberg, New York. p. 47-59.
21.
Gottesman, I.I., Shields, J., A
polygenic theory of schizophrenia. Proc
Natl Acad Sci USA, 1967. 58: p.
199-205.
22. Constantinidis, J.K., Les Marqueurs de chromosomes chez les schizophrenes et la recherche du linkage entre ces caracteres et la schizophrenie par la methode de Penrose. J Genet hum, 1958. 7: p. 189-242.
23. Turner, W.J., Genetic markers for schizotaxia. Biol Psychiatry, 1979. 14: p. 177-206. [PubMed]
24. Coon, H., et al., Genomic scan for genes predisposing to schizophrenia. Am J Med Genet, 1994. 54: p. 59-71. [PubMed]
25.
Moises, H.W., et al., An
international two-stage genome-wide search for schizophrenia susceptibility
genes. Nat Genet, 1995. 11: p. 321-324.
26. Wiese, C., Systematische genomweite Suche nach zur Schizophrenie prädisponierenden Genen. PhD Thesis, Faculty of Natural Sciences. 1994, Christian-Albrechts University at Kiel: Kiel, Germany. p. 106.
27. Pulver, A.E., et al., Schizophrenia: a genome scan targets chromosomes 3p and 8p as potential sites of susceptibility genes. Am J Med Genet, 1995. 60: p. 252-260. [PubMed]
28. SCLG 6 AND 8, Additional support for schizophrenia linkage on chromosomes 6 and 8: a multicenter study. Schizophrenia Linkage Collaborative Group for Chromosomes 3, 6 and 8. Am J Med Genet, 1996. 67: p. 580-594. [PubMed]
29. Kendler, K.S., et al., Evidence for a schizophrenia vulnerability locus on chromosome 8p in the Irish Study of High-Density Schizophrenia Families. Am J Psychiatry, 1996. 153: p. 1534-1540. [PubMed]
30. Blouin, J.L., et al., Schizophrenia susceptibility loci on chromosomes 13q32 and 8p21. Nat Genet, 1998. 20: p. 70-73. [PubMed]
31. Kaufmann, C.A., et al., NIMH Genetics Initiative Millenium Schizophrenia Consortium: linkage analysis of African-American pedigrees. Am J Med Genet, 1998. 81: p. 282-289. [PubMed]
32. Shaw, S.H., et al., A genome-wide search for schizophrenia susceptibility genes. Am J Med Genet, 1998. 81: p. 364-376. [PubMed]
33. Brzustowicz, L.M., et al., Location of a major susceptibility locus for familial schizophrenia on chromosome 1q21-q22. Science, 2000. 288: p. 678-682. [PubMed]
34. Pulver, A.E., et al., Genetic heterogeneity in schizophrenia: stratification of genome scan data using co-segregating related phenotypes. Mol Psychiatry, 2000. 5: p. 650-653. [PubMed]
35. Gurling, H.M., et al., Genomewide genetic linkage analysis confirms the presence of susceptibility loci for schizophrenia, on chromosomes 1q32.2, 5q33.2, and 8p21-22 and provides support for linkage to schizophrenia, on chromosomes 11q23.3-24 and 20q12.1-11.23. Am J Hum Genet, 2001. 68: p. 661-673. [PubMed]
36. Gottesman, I.I., Moises, H.W. Linkage findings support the multigene (QTL) theory of schizophrenia. Paper presented at The VIIth World Congress of Psychiatric Genetics. 1999. Monterey, California, USA. Mol Psychiatry, 1999. 4 (Suppl. I): p. S3 (Abstract)
37. American Psychiatric Association, Diagnostic and statistical manual of mental disorders. 4th Ed. ed. 1994, Washington DC: American Psychiatric Association.
38. Edwards, J.H., Sib-pairs in multifactorial disorders - The sib-similarity problem. Submitted, 2002.
39. Falk, C.T., Rubinstein, P., Haplotype relative risks: an easy reliable way to construct a proper control sample for risk calculations. Ann Hum Genet, 1987. 51: p. 227-233. [PubMed]
40. Sham, P.C., Curtis, D., Monte Carlo tests for associations between disease and alleles at highly polymorphic loci. Ann Hum Genet, 1995. 59: p. 97-105. [PubMed]
41. Woolf, B., On estimating the relation between blood group and disease. Ann Hum Genet, 1955. 19: p. 251-253.
42. Abecasis, G.R., Cookson, W.O.C., GOLD - Graphical Overview of Linkage Disequilibrium. Bioinformatics, 2000. 16: p. 182-183.
43. Sobel, E., Lange, K., Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker sharing statistics. Am J Hum Genet, 1996. 58: p. 1323-1337. [PubMed]
44. Chalifa-Caspi, V., Prilusky, J., Lancet, D., The Unified Database. World Wide Web URL: http://bioinformatics.weizmann.ac.il/udb. 1998, Rehovot, Israel: Weizmann Institute of Science, Bioinformatics Unit and Genome Center.
45. Burden, S., Yarden, Y., Neuregulins and their receptors: a versatile signaling module in organogenesis and oncogenesis. Neuron, 1997. 18: p. 847-855. [PubMed]
46. Buonanno, A., Fischbach, G.D., Neuregulin and ErbB receptor signaling pathways in the nervous system. Curr Opin Neurobiol, 2001. 11: p. 287-296. [PubMed]
47. Moises, H.W.M., Human Genome data analyzed by an evolutionary method suggests a decrease in protein-synthesis rate as cause of schizophrenia and an increase as antipsychotic mechanism. E-Print archive, http://arXiv.cornell.edu/abs/cond-mat/0110189, 2001.
48. Bracha, H.S., et al., Second-trimester markers of fetal size in schizophrenia: a study of monozygotic twins. Am J Psychiatry, 1992. 149: p. 1355-1361. [PubMed]
49. Wolpowitz, D., et al., Cysteine-rich domaine isoforms of the neuregulin-1 gene are required for maintenance of periphereal synapses. Neuron, 2000. 25: p. 79-91. [PubMed]
50. Mathalon, D.H., et al., Progressive brain volume changes and the clinical course of schizophrenia in men: a longitudinal magnetic resonance imaging study. Arch Gen Psychiatry, 2001. 58(2): p. 148-157. [PubMed]
51. Lieberman, J., et al., Longitudinal study of brain morphology in first episode schizophrenia. Biol Psychiatry, 2001. 49: p. 487-499. [PubMed]
52. Meltzer, H.Y., Creatine kinase and aldolase in serum: abnormality common to acute psychoses. Science, 1968. 159: p. 1368-1370. [PubMed]
53. Meltzer, H.Y., Muscle enyzme release in the acute psychoses. Arch Gen Psychiatry, 1969. 21: p. 101-112. [PubMed]
54. Crayton, J.W. and H.Y. Meltzer, Motor endplate alterations in schizophrenic patients. Nature, 1976. 264: p. 658-659. [PubMed]
55. Crayton, J.W. and H.Y. Meltzer, Degeneration and regeneration of motor neurons in psychotic patients. Biol Psychiatry, 1979, 14:803-819. [PubMed]
56. Goode, D.J., et al., Physiologic abnormalities of the neuromuscular system in schizophrenia. Schizophr Bull, 1977. 3: p. 121-138. [PubMed]
57. Ross-Stanton, J. and H.Y. Meltzer, Motor neuron branching patterns in psychotic patients. Arch Gen Psychiatry, 1981. 38: p. 1097-1103. [PubMed]
58. Huang, Y.Z., et al., Regulation of neuregulin signaling by PSD-95 interacting with erbB4 and CNS synapses. Neuron, 2000. 26: p. 443-455. [PubMed]
59. Orban, P.C., P.F. Chapman, and R. Brambilla, Is the Ras-MAPK signalling pathway necessary for long-term memory formation? Trends Neurosci, 1999. 22: p. 38-44. [PubMed]
60. Mazzucchelli, C. and R. Brambilla, Ras-related and MAPK signalling in neuronal plasticity and memory formation. Cell Mol Life Sci, 2000. 57: p. 604-611. [PubMed]
61. Gold, J.M., et al., Learning and forgetting in schizophrenia. J Abnorm Psychol, 2000. 109: p. 534-538. [PubMed]
62. Saykin, A.J., et al., Neuropsychological function in schizophrenia. Selective impairment in memory and learning. Arch Gen Psychiatry, 1991. 48: p. 618-624. [PubMed]
63. Heinrichs, R.W. and K.K. Zakzanis, Neurocognitive deficit in schizophrenia: a quantitative review of the evidence. Neuropsychology, 1998. 12: p. 426-445. [PubMed]
64. Brebion, G., et al., Memory impairment and schizophrenia: the role of processing speed. Schizophr Res, 1998. 30: p. 31-39. [PubMed]
65. Frith, C., Dolan, R.J., The Role of Memory in the Delusions Associated with Schizophrenia, in Memory, Brain, and Belief, D.L. Schacter, Scarry, E., Editor. 2000, Harvard University Press: Cambridge, MA. p. 115-135.
66. Frith, C.D., S. Blakemore, and D.M. Wolpert, Explaining the symptoms of schizophrenia: abnormalities in the awareness of action. Brain Res Brain Res Rev, 2000. 31(2-3): p. 357-363. [PubMed]
67. Hynes, N.E., Stern, D.F., The biology of erbB-2/neu/HER-2 and its role in cancer. Biochim Biophys Acta, 1994. 1198: p. 165-184. [PubMed]
68. Tang, C.K., Lippman, M.E., EGF family receptors and their ligands in human cancer, in Hormones and Signaling, B.W. O'Malley, Editor. 1998, Academic Press: San Diego, CA. p. 113-165.
69. Dupont, A., Moeller-Jensen, O., Incidence of cancer in patients diagnosed as schizophrenic in Denmark, in Psychiatric case registers in public health, G.H. ten Horn, Giel, R., Gulbinat, W., Henderson, J.H., Editor. 1986, Elsevier: Amsterdam. p. 229-239.
70. Saku, M., et al., Mortality in psychiatric patients, with a specific focus on cancer mortality associated with schizophrenia. Int J Epidemiol, 1995. 24: p. 366-372. [PubMed]
71. Hakak, Y., et al., Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci U S A, 2001. 98: p. 4746-4751
72. Pinkas-Kramarski, R., et al., ErbB tyrosine kinases and the two neuregulin families constitute a ligand-receptor network. Mol Cell Biol, 1998. 18: p. 6090-60101. [PubMed]
73. Hertig, C.M., Kubalak, S.W., Wang, Y., Chien, K.R., Synergistic roles of neuregulin-1 and insulin-like growth factor-I in activation of the phosphatidylinositol 3-kinase pathway and cardiac chamber morphogenesis. J Biol Chem, 1999. 274: p. 37362-3736.
74. Ehrlich, S., Goldshmit, Y., Lupowitz, Z., Pinkas-Kramarski, R., ErbB-4 activation inhibits apoptosis in PC12 cells. Neuroscience, 2001. 107: p. 353-362. [PubMed]
75. Alroy, I., Yarden, Y., The ErbB signaling network in embryogenesis and oncogenesis: signaling diversification through combinatorial ligand-receptor interactions. FEBS Lett, 1997. 410: p. 83-86. [PubMed]
76. Eilam, R., et al., Activity-dependent regulation of Neu differentiation factor/neuregulin expression in rat brain. Proc Natl Acad Sci U S A, 1998. 95: p. 1888-1893.