To
cite this article use: Clin Med Health Res (2001) clinmed/2001110002
Copyright © 2001 Alexei
Koudinov and Natalia Koudinova
AMYLOID PLAQUE ( AND NOT DIFFUSE
AMYLOID ) IS A CONDITION FOR NEURONAL DYSFUNCTION
Alexei R. Koudinov,a,CA
Temirbolat T. Berezova and Natalia V. Koudinovaa,b
a Berezov Academic Laboratory, Russian Academy of Medical
Sciences, Timoshenko St., 38-27, Moscow, 121359 Russia;
b Weizmann Institute of Science, Department of Biological
Regulation, Rehovot, 76100 Israel.
First submitted: December
23, 1999, Published online: December, 2001
ARTICLE NAVIGATION MENU
Please remember about your browser
shortcuts: to navigate back and forth use <CTRL><LEFT> and <CTRL><RIGHT>
keys on your keyboard.
ABSTRACT 
There is no direct
evidence that brain amyloid affects neuronal function. In this report
we studied hippocampal slices from non-mutated human amyloid precursor
protein (APP695)ABBR
transgenic- and age-matched non-transgenic control mice. We aimed to differentiate
separate actions of the aged (25.5 months) transgenic mice plaque-like
amyloid and diffuse amyloid of the non-transgenic mice (verified by immunohistochemistry
and Congo Red fluorescence) on synaptic plasticity. Extracellular recording
of CA1 field excitatory postsynaptic potentials in vitro revealed impairment
of input/output characteristics, long-term potentiation, and the delay
of few milliseconds in initial post-tetanic traces in aged transgenic versus
control mice hippocampal slices. Our results indicate that amyloid plaque (and not
diffuse amyloid) may cause synaptic dysfunction, and suggest importance factors other then brain amyloid in pre-plaque stages of Alzheimers disease and in Down syndrome.
INTRODUCTION
Diffuse amyloid
deposits and neuritic plaques of Alzheimer's disease patients are considered
to be essential disease features [ 1 ] For this
reason the prevention of amyloid formation from its precursor (APP)
and inhibition of amyloid fibrillogenesis have been proposed as an important
therapeutic targets for the disease cure [ 2, 3,
4
]. Nevertheless, there is no direct evidence that amyloid b
(Ab) has direct effect on neuronal dysfunction.
An attempt to unravel this important issue was made in a report on transgenic
mice expressing human amyloid precursor protein (APP695) bearing
the swedish mutation [ 5 ]. These transgenic mice developed
"elevated concentrations of Ab and significant
amyloid deposits," and had impaired spatial learning and hippocampal long
term potentiation (LTP), a long-lasting synaptic enhancement, the leading
experimental model for the synaptic changes that underlie learning and
memory [ 6WEB+ ]. However, cited report [ 5
] as well as another earlier work on LTP deficit in transgenic mice overexpressing
the carboxy-terminal 104 aminoacids of APP [ 7 ], did
not "determine whether the effects measured resulted from elevated concentrations
of soluble Ab, deposited Ab
or both".
Todate, it is not
established whether the maturation of brain amyloid deposits, particularly
the development of Congo Red positive neuritic plaques, is an essential
event leading to neuronal dysfunction. Recent study by Naslund et
al. [ 8 ] attempted to correlate the amyloid load
with the cognitive decline and the severity of dementia in Alzheimer disease
patients. However, the latter report estimated 55% and 40% of the study
subjects without detectable plaques as the ones having readily detectable
levels of "Ab(x-42)
and Ab(x-40)" .
In addition, recent reports have suggested the possible importance of factors
other then brain amyloid in Alzheimer's neuronal abnormality (oxidative
stress [ 9WEB+ ] or lipid metabolism [ 10WEB+,
11
] disbalance, for example) and behavioral disturbances without amyloid
deposits in mice overexpressing human APP with Flemish and Dutch mutations
[ 12 ], keeping open the question on the role of neuritic
plaques in the genesis of Alzheimer's neuronal dysfunction.
In our study, we utilized hippocampal slices of
16.5 and 25.5 month old transgenic mice expressing nonmutated human APP695
[ 13 ], and age-matched non-transgenic wild type control
mice. We aimed to differentiate the separate actions of diffuse and plaque
amyloid on hippocampal synaptic plasticity using immunohistochemical analysis,
Congo Red staining, amyloid extraction and extracellular recording of CA1
field excitatory postsynaptic potentials ( fEPSPs ). The results indicate
that amyloid plaque (and not diffuse amyloid) may represent one of the
possible causes of neuronal dysfunction and synaptic plasticity failure.
EXPERIMENTAL
PROCEDURES
Animal care and tissue collection
Transgenic and wild type mice of 16.5 and 25.5 months age groups
(n=6) were maintained on the standard diet at the animal facility of the
Weizmann Institute of Science, Rehovot, Israel. All experimental
procedures were in accord with National Institutes of Health guide for
the use of laboratory animals. Two or three hippocampal slices from each
mouse were subjected to both electrophysiology and to immunohistochemistry.
Additionally, two transgenic and two wild type mice at the age of 16.5
months were subjected to transcardial perfusion, followed by thin sectioning
of the brain and immunohistochemical analysis. In selected experiments,
the hippocampal slices of 16.5 month old transgenic and wild type
mice were subjected to the biochemical analysis of Ab.
Ex-vivo hippocampal slices
Hippocampal slices were prepared and electrophysiological analysis was
performed essentially as we described previously [ 10,
14
]. Briefly, after mice decapitation the hippocampus was rapidly
removed and placed into cold (20C) artificial
cerebrospinal fluid (ACSF, pH 7.4 containing (in mM): 124 NaCl, 2.0 KCl,
1.24 KH2PO4,
2.0 MgSO4, 2.5 CaCl2,
26 NaHCO3 and 10 D-glucose)
saturated with 95 % O2
/ 5% CO2 (flow rate
0.4 l/min) and adjusted with sucrose (7g per 600 ml of ACSF) to 320 mOsm
osmolarity. The hippocampal slices (400 m)
were prepared with a McIlwain tissue slicer, USA. The slices were incubated
in a recreation chamber at room temperature (250C)
for 1.5 h in ACSF.
Electrophysiology
The slices were transferred to a recording chamber (held at constant
temperature of 320C) and submerged slices
were superfused with ACSF at a flow rate of ~1.5 ml/min. Extracellular
electrodes (~4 MW, 0.75 mM NaCl) were guided
by micromanipulator into stratum radiatum of CA1 (200 m deep) under binocular.
Bipolar (A. Koudinov fabricated) tungsten (50 m
wire size) stimulating electrode was also placed into CA1 stratum radiatum.
The stimulations were delivered every 30 s at 50 ms
pulse duration yielding fEPSP waveforms. After stable baseline responses
were established and input /output (I/O) curve values recorded, tetanus
induced LTP was induced by delivering a 100 Hz, 1 sec stimuli train through
the stimulation electrode at the baseline test stimulus intensity. For
long term depression (LTD) study slices were stimulated every half a second
(2 Hz) for 5 min at the test stimulus intensity.
Data were collected, stored and analyzed on a PC using Asyst 3.1 and
GraphPad Prizm 2.0 data acquisition and analysis software. The I/O relationship,
LTP and LTD were expressed as a fEPSP amplitude and slope change versus
stimulus intensity and time, respectively. Data were normalized with respect
to the steady baseline values and expressed as mean±SEM. Non-parametric
unpaired Mann-Whitney test was used for determining significant differences
between potentiation/depression levels of trangenic and wild type slices
at the indicated time points. A probability of 0.05 (one tailed) or less
was accepted as statistically significant.
Immunohistochemistry and Congo Red staining
for amyloid
The hippocampal slices from 25.5 month old mice used for synaptic plasticity
study, were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS),
pH=7.4 for 72 hrs [ 10, 14 ].
We also used hippocampal sections from 16.5 month old mice killed by transcardial
perfusion with PBS and 4% paraformaldehyde in PBS [ 11,
14
]. For immunohistochemistry, 400 m-thick fixed
slices were cutted with a microtome to a 40 m
sections. Free floating sections were washed in PBS (2x) and then incubated
with 3% H202 (prepared on PBS, containing 10% methanol) for 20 min to remove
endogenous peroxidase activity. Sections were then washed (4x 10 min each)
with PBS and blocked for 24 hrs at 40C
in 10% fetal calf serum and 1% glycine in PBS (blocking solution), followed
by 14 hrs incubation with 4G8 or 6E10 (1:1000, Senetek, PLC.) monoclonal
anti-Ab antibodies in the blocking solution
at 40C. Tissue sections were washed (5x
40 min each) in blocking solution to remove unreacted primary antibodies.
Secondary biotinylated goat anti-mouse antibody (1:750) were added for
1.5 h at 250C, followed by washing with
the blocking buffer (3x 40 min each). After washing, ABC solution
(Vector Elite kit, 1:300 of reagents A and B in blocking solution) was
added for 40 min, followed by section washing in blocking solution and
then in PBS (2x 10 min each). For visualization, immunostained sections
were reacted with 3',3-diaminobenzidine tetra-hydrochloride (Sigma Tablet
kit, USA) and washed in PBS.
For Congo Red staining thin sections were stained with 0.5% Congo Red
in 50% alcohol for 30 min, followed by one minute treatment with 0.2% KOH
in 80% alcohol and water. Sections were mounted on gelatinized slides,
air dried, dehydrated in serially diluted ethanol (50, 70, 90, 95 and 100%),
cleared with Xylenes, and coverslipped with cover glass and Permount, USA.
To control the specificity of 4G8 and 6E10 immunostaining, antibody
solutions were preadsorbed with the access of synthetic peptide Ab1-40
(1 mg /0.5 ml) prior to the incubation with the sections.
Fluorescence of plaque-like amyloid labeled by Congo Red was obtained
with 488 nm of excitation using a confocal microscope LSM 510 (Zeiss, Germany)
equipped with an argon laser [ 1S
].
Amyloid extraction
Hippocampal slices were homogenized with the cold PBS (0.5 ml
per 20 mg of tissue) containing protease inhibitors [ 15
], and subjected to centrifugation in a Beckman TiL 100.2 rotor at 100,000
g for 3 hrs at 40C. The supernatant fraction
and the pellet were dialyzed against water with 1,000 Da cut-off membrane
(Spectrum, USA), lyophilized and subjected to Ab
extraction with 20% and then with 80% acetonitrile in 0.1% trifluoroacetic
acid [ 16 ]. Both acetonitrile soluble fractions were
combined, lyophilized and subjected to 13% TRIS-Tricine SDS/PAGE [ 17,
18
] and immunoblot analysis on Immobilon P membranes (Wattman, USA) with
6E10 and 4G8 anti-Ab monoclonal antibodies and
with the monoclonal antibody against APP (Zymed, USA), followed by ECL
(Amersham) essentially as previously reported [ 15,
18
].
RESULTS
AND DISCUSSION
A. IMMUNOHISTOCHEMICAL
ANALYSIS: AMYLOID AS ALZHEIMERS HALLMARK
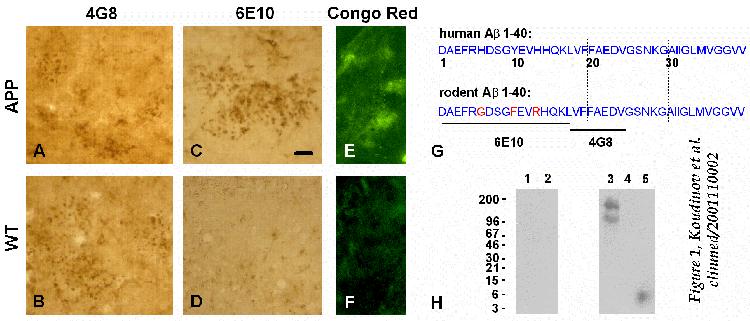
Immunohistochemistry
of slices from aged animals (25.5 months) with 4G8 and 6E10 antibodies
(anti-human/mouse-Ab and anti-human-Ab,
respectively, see antibody specificity scheme, Figure
1G ) revealed extracellular (staining with no triton) hippocampal
immunoreactivity of mouse Ab in transgenic mice
(Fig. 1A) and in wild types (Fig. 1B),
and verified extracellular deposits of human Ab
in the transgenic mice hippocampus (Fig. 1C). Essentially
identical results were obtained under the condition of membrane permeabilization
with 0.1% Triton X-100 in the blocking buffer (not shown). The specificity
of immunostaining was confirmed by the preadsorption of antibodies with
the access of synthetic peptide Ab(1-40).
Staining for amyloid showed Congo Red fluorescence specifically in aged
transgenic mice (and not in aged wild type mice) hippocampal sections
(Fig. 1E). Congo red stains specifically amyloid plaques
(but not diffuse amyloid) due to the binding to the b-pleated
sheet secondary structure of Ab protein in amyloid
fibrils [ 1, 2 ]. The latter observation
indicates that expression of non-mutated human Ab
in aged transgenic mice leads to a mature Alzheimer's plaque-like amyloid
and that Ab deposits in wild type mice
have a nonmature diffuse nature.
In contrast to the 25.5 month aged animals, the 16.5 month old transgenic
and wild type mice expressed neither human nor mouse Ab
immunoreactivity (not shown). To confirm this observation we analyzed 16.5
month old transgenic and wild type mice for soluble and aggregated Ab
by immunoblot analysis of the acetonitrile extracts of ultracentrifugation
supernatant- and pellet-fractions of hippocampal homogenates. We did not
recover human transgenic (Fig. 1H) or mouse (not shown)
Ab immunoreactivity in the transgenic and wild
type hippocampus. However, anti-human Ab antibody
6E10 labeled a protein of high molecular weight (in the range of 96 to
200 kDa) in the trangenic mice. This protein band was also stained with
the antibody against APP aminoterminus (not shown) confirming human APP
expression in the transgenic hippocampus.
FIGURE 1
 You may need to resize your browser
window for better figure view.
You may need to resize your browser
window for better figure view.
Immunochemical analysis of APP transgenic mice.
A-D, Comparison of extracellular Ab
immunoreactivity in aged (25.5 months) human non-mutated APP695
transgenic mice and age-matched non-transgenic wild type control mice hippocampus.
Immunohistochemistry was performed with no triton and 4G8 and 6E10 monoclonal
antibodies (1:1000). The presented fields are CA1 areas of ex-vivo
slices from the batches used for extracellular recording; Bar, 20
m.
Similar Ab pattern was observed in the dentate
gyrus (not shown). Alzheimer's-like plaque amyloid in transgenic mice (E),
versus wild type mice (F), was visualized by confocal fluorescent
microscopy of Congo Red stained sections; Bar, 100 m.
(H) 16.5 month old wild type (control, lanes 1 and 2) and transgenic
mice (lanes 3 and 4) were analyzed for the soluble and/or aggregated human
Ab in the acetonitrile extracts of the supernatant
(lanes 1 and 3) and pellet (lanes 2 and 4) fractions of the hippocampal
homogenates, respectively, by immunoblot analysis with 6E10 and 4G8 (not
shown) monoclonal antibody. While there were no detectable levels of Ab
present in transgenic hippocampus, we recovered 6E10-positive high molecular
weight human APP immunoreactivity in the supernatant fraction (lane 3),
and confirmed it by immunostaining with the antibody against APP aminoterminus.
Lane 5, 5 ng of synthetic Ab1-40 (positive control).
Molecular weight markers (in kDa) are shown on the left. Scheme (G)
represents human and rodent Ab1-40 amino acid
sequence differences and the sequence specificity of anti-Ab
monoclonal antibodies used in this study.
B. ELECTROPHYSIOLOGICAL
ANALYSIS: PLAQUE AMYLOID AND SYNAPTIC PLASTICITY
Electrophysiological
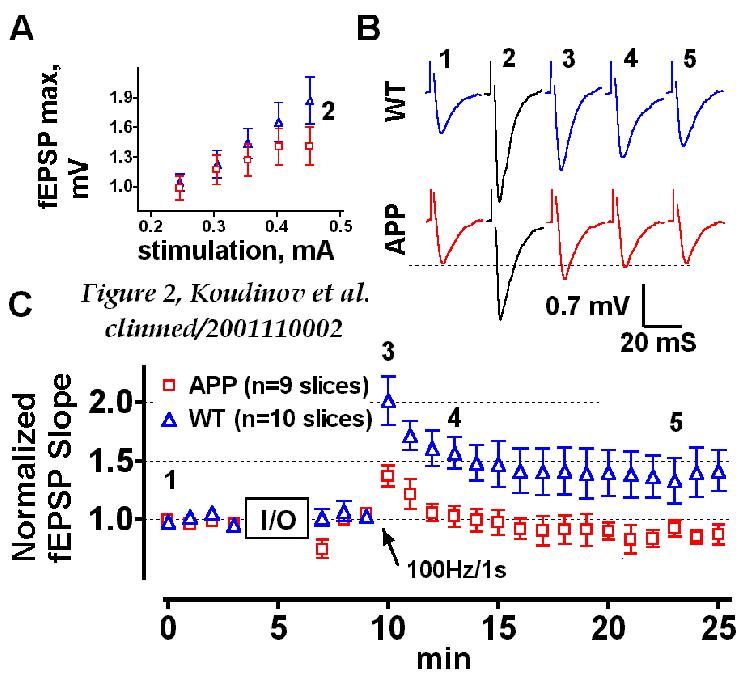
analysis revealed that 25.5 month old transgenic mice had lower input-stimulus/output-response
(I/O) characteristics (Figure 2A). It is generally
accepted that in the CA1 area of the hippocampus EPSP consists of two major
elements that depend on the activation of N-methyl-D-aspartate (NMDA) and
non-NMDA (mainly AMPA) subtypes of ionotropic glutamate receptors [ 19WEB+
]. The 50 ms stimulation pulse duration (employed
here to evoke fEPSP) generates significant AMPA responses and it is thus
possible that this particular component of the EPSP is responsible for
lower I/O characteristics of aged transgenic mice [ 19WEB+
].
Aged transgenic mice were impaired in both the amount of initial post-tetanic
potentiation (137.2±9.3%, n=9, against 201.8±20.8% of the
wild type, n=10, p=0.014) and in the maintenance of the LTP, sampled 4
smin (99.53±9.78%, n=9, and 148.6±15.01%, n=10, p=0.0318)
and 10 min (92.64±7.02%, n=9, and 133.2±9.29%, n=10, p=0.0357)
after the tetanic stimulation. There were no significant differences
in induction (p=0.2403) and maintenance (p=0.0649) of the LTP in wild type
slices taken from 16.5 and 25.5 months old mice (Fig. 2
and Figure 3A) despite the development of diffuse
mouse Ab deposits in wild type hippocampus over
the indicated age (indicating the lack of importance of diffuse amyloid
for synaptic plasticity failure). On the other hand, transgenic slices
expressing plaque-like amyloid at age 25.5 months showed a significant
decline in both induction (p=0.0047) and maintenance (p=0.0048) of the
LTP compared to the 16.5 month old mice.
FIGURE 2
 You may need to resize your browser
window for better figure view.
You may need to resize your browser
window for better figure view.
Electrophysiological analysis of aged (25.5 months) APP transgenic
mice
(A) Input/output (I/O) relationship in APP transgenic
(squares) and wild type non-transgenic (WT, triangles) hippocampal slices.
(B) Field synaptic responses obtained at the baseline (1) and high
stimulus intensity (2) recordings as well as immediately after (3) and
3 (4) and 13 min after (5) the high-frequency train of stimuli. (C)
Impairment of tetanic LTP in CA1 of ex-vivo slices from APP transgenic
versus WT hippocampus. In all cases n=9 and n=10 slices for APP transgenic
and WT mice, respectively. Arrow indicates time of tetanus.
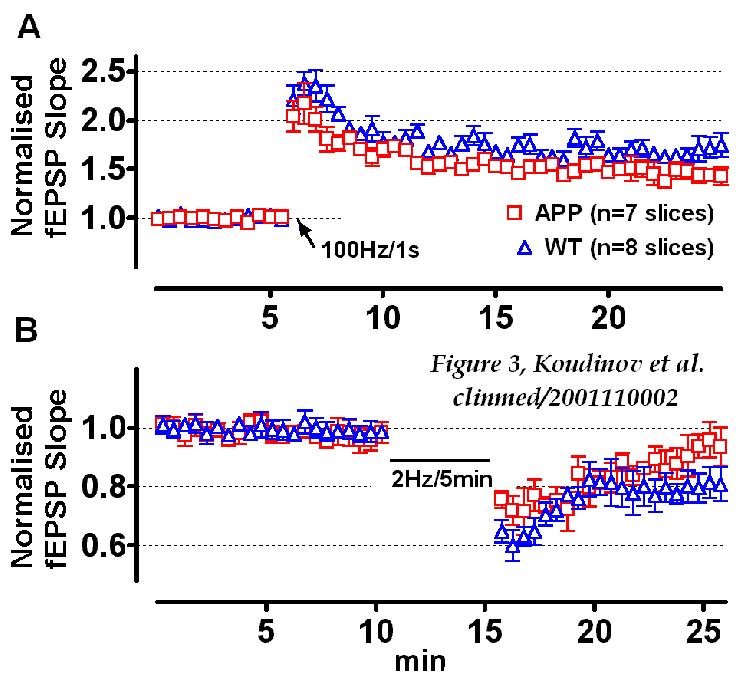
In contrast to the aged 25.5 month old mice, transgenic mice of the
younger age (16.5 months) expressed no amyloid and did not differ from
the wild type mice in the induction and maintenance of the LTP (Fig. 3A).
There were, however, differences in the LTD (Fig. 3B), another important
parameter of neuronal plasticity. It was shown previously [ 20
] that bath application of the soluble APP (100 nM, 1 h) adsorbed the ability
of rodent hippocampal slices to maintain LTD. It is thus conceivable that
mild modulation of the hippocampal LTD in adult transgenic mice (16.5 months)
is due to the human APP expression in the transgenic mice hippocampus (Fig. 1H). Another study by Larson et al. [ 21
], however, suggests a more striking modulation of hippocampal physiology
by human mutant (V717F)APP in transgenic mice at age 4-5 months. Although
the experimental protocol of this report (specifically, maintaining slices
at 360C; differences in the media recipe,
particularly including ascorbate, known to modulate EPSP [ 22
] in ACSF) does
not match the one employed here and in the above cited report by Ishida
et al. [ 20 ], it indicates that APP mutations
(yet representing very small cohort of all Alzheimer disease cases) may
exacerbate additional abnormalities in synaptic and behavioral plasticity
"prior to the formation of amyloid beta peptide deposits." The transgenic
mice that we used in this study mild overexpress non-mutated human APP
and in our view offer more relevant system to model non-mutated human
APP expression [ 13 ].
FIGURE 3
 You may need to resize your browser
window for better figure view.
You may need to resize your browser
window for better figure view.
Electrophysiological analysis of 16.5 month old APP transgenic
mice
APP transgenic and wild type non-transgenic (WT) mice
expressed similar tetanus (arrow) induced LTP (A) and were different
in the amount of the long term depression (B). However, depression
values probed 10 min after the low frequency stimulation sequence did not
reach statistical significance (98.95±10.34%, n=7, and 79.3±12.8%,
n=6 in the APP-TG and WT, respectively, p=0.0625, one-tailed).
Two major components contributing to the tetanus induced LTP are NMDA
LTP and non-NMDA (dependent on a rise in intracellular calcium concentration)
LTP [ 23WEB+ ]. Fast onset NMDA- and developing slow
non- NMDA-LTP can be isolated by using a specific tetanic stimulation paradigm
in the presence of 30 mM nifedepine, a blocker
of voltage-gated calcium channels, and 25 mM
D,L-2-amino-5-phosphonovaleric acid, an NMDA antagonist, respectively [
23WEB+,
24
]. Moreover, another type of slow onset LTP was described, a muscarinic
LTP, which can be evoked by the application of 0.25-2.0 mM
carbachol in the absence of tetanic stimulation [ 23WEB+,
24
]. Non-NMDA-LTP and muscarinic LTP share similar lack of expression in
adult transgenic mice expressing human Cu/Zn-superoxide dismutase, SOD1
[ 24 ] in very old (24-30 months) and in adult Wistar
rat hippocampal slices treated with low (~30
mM)
dose of H2O2
[ 23 ].
Regardless of the deficit of specific receptor machinery hippocampal
slices from aged transgenic mice may be different from wild type controls
in their ability to regulate second messenger pathways and/or generate
the action potential. Thus, transgenic mice may be impaired in the
phosphorylation of the nuclear cAMP responsive element binding protein
(abbreviated as CREB), modulated by micromolar concentrations of Ab
[ 25 ] and known as a necessary event in neuronal
plasticity [ 26 ]. Transgenic mice may be also
impaired in metabotropic glutamate receptors (mGluR) and associated second
messenger machinery, activation which was shown to be coupled to the APP
processing [ 27 ] and is essential for the priming
of the LTP [ 28 ]. It is also possible that in transgenic
mice neurons are depolarized relative to the control slices, yielding reduction
of their fEPSPs and impairment of their ability to express larger fEPSPs
following tetanic stimulation.
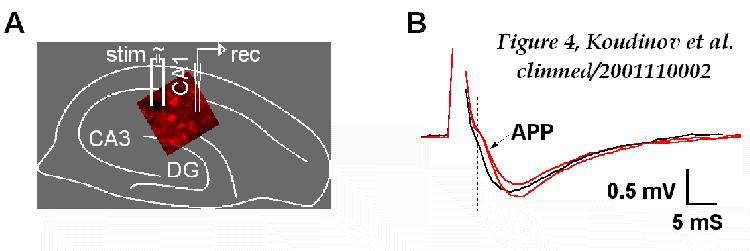
Finally, transgenic mice may have deteriorated neuronal spike propagation
machinery due to the tunneling amyloid plaques (Figure
4A). This is supported by the study on the disruption of neural
networks in Alzheimer's disease [ 29 ]. This report
modeled the electrophysiological effect of the changed neuronal processes
that cross through Ab plaque deposit and foretold
the delay of several milliseconds over an average plaque. Our comparison
of individual fEPSPs revealed this predicted change of few milliseconds
in initial post-tetanic traces in transgenic versus wild type slices
(Fig. 4B), confirming importance of plaque amyloid
for neuronal spike propagation.
FIGURE 4
 You may need to resize your browser
window for better figure view.
You may need to resize your browser
window for better figure view.
Schematic matching of the positioning of the recording (rec) and
stimulating (stim) electrodes
for the employed in the study extracellular recording
in the CA1 and the Congo Red fluorescence, observed in the APP transgenic
hippocampal slice (A). (B) Individual fEPSPs recorded after
the high frequency (100 Hz/1 s) train of stimuli revealed 1.5-2.0 msec
delay in the onset of the evoked synaptic responses in the APP transgenic
(arrow) compared to the wild type non-transgenic slices.
CONCLUSION
Our data provide evidence that one of the possible causes of Alzheimer's-like
neuronal dysfunction and synaptic plasticity deficit is senile plaque (and
not diffuse) amyloid. Our data are in dispute with the paper
in JAMA [
8 ] and the accompanying commentary [ 30WEB+
, see also 31WEB+ ] proposing that Ab
peptide not incorporated into histologically visible plaques is an early
neurochemical hallmark of dementia [ 2S,
3S
].
Our results also suggest that in Down syndrome (characterized by
diffuse amyloid deposition in early life) and in pre-plaque stages of Alzhemer
disease, the other factors (such as brain cholesterol and other lipid metabolism
misregulation [ 10WEB+, 11
,
LE1,
LE2,
LE3,
LE4,
LE5
] or oxidative stress disbalance [ 9WEB+,
24
]) may contribute to the neuronal and behavioural [ 2S,
3S
] dysfunction.
The precise molecular mechanism of amyloid-plaque-mediated synaptic
plasticity deficit, however, remains to be investigated.
ACKNOWLEDGMENT
While this paper was under submission another key contribution
on this subject was published in December 2000 [ 4S
]. We are indebted to Sh. Halav and J. Hermesh for excellent animal care.
See Ref. 10 for support and funding itemization. The
preliminary account of this report has been published in the abstract form
and presented at the 10th Meeting of the European Neurological Society,
18-21 June 2000, Jerusalem, Israel [ 5S ], and
at World Alzheimer congress 2000, Washington, DC, July 9-12, 2000 [ 6S
]. The earlier version of this article was first submitted as research
letter (for letter text see [ 5S ]) during December
23, 1999. In the present form this contribution was submitted to Science,
J
Neurosci and to Neurosciences. Our research is dedicated to
our parents.
MISCELLANEA
| FOR
YOUR CONVENIENCE:
|
THIS
ARTICLE IS BASED ON:
 |
Authors near 10 years expertise on the synthesis of research
on soluble Ab, lipids and Alzheimer's disease,
and 5 years of basic neuroscience research. |
|
31 web references, 18 WEB+ citations, plus supplementary
references. |
|
KEY
TAKE-HOME MESSAGES
|
Todate, it is not established whether
the maturation of brain amyloid deposits, particularly the development
of amyloid neuritic plaques, is an essential event leading to neuronal
dysfunction in Alzheimer's patients. |
|
Our data provide evidence that
amyloid plaque (and not diffuse amyloid) impairs neuronal function and synaptic plasticity. |
|
Our current results and recent
discussion suggest that in pre-plaque stages of Alzhemer disease
and in Down syndrome other factors (such as brain cholesterol/lipid
metabolism misregulation or oxidative stress disbalance)
have primary causative role for the development of neuronal dysfunction.
Amyloid plaque buildup is not causative for the sporadic Alzheimer's disease
and unlikely represents the proper target for Alzheimer's therapy. |
|
FOOTNOTES
|
CACorresponding author: Alexei
R. Koudinov, M.D., Ph.D., P.O.Box 1665, Rehovot 76100 Israel; Tel:
(972 8) 947-1803; Fax: (972 8) 934-4116; E.contact: amyloidbeta@hotmail.com
; koudin@imb.ac.ru ; Internet
Office |
|
ABBRAbbreviations:
AD,
Alzheimer's disease; APP, amyloid precursor protein; Ab,
amyloid b protein; DS, Down syndrome; LTP, long-term
potentiation; LTD, long-term depression; NMDA, N-methyl-D-aspartate;
PBS, phosphate buffered saline; SOD, superoxide dismutase; TG, transgenic
mice; WT, wild-type non-transgenic mice. |
|
Keywords: Alzheimer's disease, amyloid beta protein
precursor, cholesterol, Down syndrome, etiology, extracellular recording,
EPSP, hippocampus, learning, lipid, long term potentiation and depression,
LTP, LTD, memory, neurodegeneration marker, neurofilament, oxidative stress
cascade, PHF NFT tau phosphorylation, phospholipids, plaque, secretase,
synaptic plasticity, therapy |
|
WEB ENHANCED
REFERENCES
1. Koudinova NV, Berezov TT,
Koudinov AR. Amyloid beta: ALzheimer's disease and brain beta amyloidoses.
Biochemistry
(Moscow) 64, 752-757 (1999) [ PubMed
] [ Reprint
Order ].
2. Koudinov AR, Koudinova NV,
Berezov TT, Ivanov YD. HDL Phospholipid: a natural inhibitor of Alzheimer's
amyloid b fibrillogenesis? Clin Chem Lab
Med. 37, 993-994 (1999) [ PubMed
] [ Abstract
] [ Reprint
Order ].
3. Schenk D, Barbour R, Dunn
W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like
pathology in the PDAPP mouse. Nature 400, 173-177 (1999)
[ PubMed
].
4. Soto C, Sigurdsson EM, Morelli
L, Kumar RA, Castano EM, Frangione B. Beta-sheet breaker peptides inhibit
fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer's
therapy. Nature Med. 4, 822-826 (1998) [ PubMed
].
5. Chapman PF, White GL, Jones
MW, et al. Impaired synaptic plasticity and learning in aged amyloid precursor
protein transgenic mice. Nature Neurosci. 2, 271-276 (1999)
[ PubMed
].
6. ( 3 web+ citations )
Rioult-Pedotti M-S, Friedman D, Donoghue JP. Learning-induced LTP in Neocortex.
Science 290,
533-536 (2000) [ PubMed
]; Bliss TVP, Collingridge JL. A synaptic model of memory: long term
potentiation in the hippocampus. Nature 361, 31-39
(1993) [ PubMed
]; Malenka RC, Nicoll RA. Long-term potentiation--a decade of progress?
Science
285,
1870-1874 (1999) [ PubMed
].
7. Nalbantoglu J, Tirado-Santiago
G, Lahsaini A, et al. Impaired learning and LTP in mice expressing
the carboxy terminus of the Alzheimer amyloid precursor protein. Nature
387,
500-505 (1997) [ PubMed
].
8. Naslund J, Haroutunian V,
Mohs R, et al. Correlation between elevated levels of amyloid b-peptide
in the brain and cognitive decline. JAMA. 283, 1571-77 (2000)
[ PubMed
].
9. ( 5 web+ citations )
Perry G, Nunomura A, Hirai K, Takeda A, Aliev G, Smith MA. Oxidative
damage in Alzheimer's disease: the metabolic dimension. Int J Dev Neurosci.
18,
417-421 (2000) [ PubMed
]; Kontush A. Amyloid-beta: an antioxidant that becomes a pro-oxidant and
critically contributes to Alzheimer's disease. Free Radic Biol Med.
31,
1120-1131 (2001) [ PubMed
]; Nunomura A, Perry G, Pappolla MA, et al. Neuronal oxidative stress precedes
amyloid-beta deposition in Down syndrome. J Neuropathol Exp Neurol.
59,
1011-1017 (2000) [ PubMed
]; Nunomura A, Perry G, Aliev G, et al. Oxidative damage is the earliest
event in Alzheimer disease. J Neuropathol Exp Neurol. 60,
759-767 (2001) [ PubMed
]; Gahtan E, Auerbach JM, Groner Y, Segal M. Reversible impairment
of long-term potentiation in transgenic Cu/Zn-SOD mice. Eur J Neurosci.
10,
538-544 (1998) [ PubMed
].
10. ( 5 web+ citations ) Koudinov
AR, Koudinova NV. Essential role for cholesterol in synaptic plasticity
and neuronal degeneration.
FASEB J. 15, 1858-1860 (2001),
published online June 27, 2001, 10.1096/fj.00-0815fje [ PubMed
Citation ] [ Full
Text ] [ Authors
Preface ]; Koudinova NV, Koudinov AR, Yavin E. Alzheimers Ab1-40
peptide modulates lipid synthesis in neuronal cultures and intact rat fetal
brain under normoxic and oxidative stress conditions. Neurochem
Res. 25, 653-660 (2000) [ PubMed
] [ Abstract
] [ Reprint
order ]; Eckert GP, Wood WG, Muller WE. Effects of aging and beta-amyloid
on the properties of brain synaptic and mitochondrial membranes. J Neural
Transm. 108, 1051-1064 (2001) [ PubMed
]; Michikawa M, Gong JS, Fan QW, Sawamura N, Yanagisawa K. A novel action
of Alzheimer's amyloid beta-protein (Abeta): oligomeric Abeta promotes
lipid release. J Neurosci. 21, 7226-7235 (2001) [ PubMed
]; Chochina SV, Avdulov NA, Igbavboa U, Cleary JP, O'Hare EO, Wood WG.
Amyloid beta-peptide(1-40) increases neuronal membrane fluidity. Role of
cholesterol and brain region. J Lipid Res. 42, 1292-1297
(2001) [ PubMed
].
11. Koudinov AR, Koudinova
NV. Brain Cholesterol Pathology is the Cause of Alzheimer's Disease. Clin
Med Health Res. published online November 27, 2001, clinmed/2001100005,
http://clinmed. netprints.org/cgi/content/full/2001100005v1 [ Full
Text ] [ Authors
Preface ] [ Letter to the Editor ].
Cited above recent key article
discusses the role for cholesterol, amyloid b
and tau in synaptic plasticity, neurodegeneration and Alzheimer's disease.
12. Kumar-Singh S, Dewachter
I, Moechars D, et al. Behavioral disturbances without amyloid deposits
in mice overexpressing human amyloid precursor protein with Flemish (A692G)
or Dutch (E693Q) mutations. Neurobiol Dis. 7, 9-22 (2000)
[ PubMed
].
13. Lamb BT, Sisodia SS, Lawler
AM, et al. Introduction and expression of the 400 kilobase precursor amyloid
protein gene in transgenic mice. Nature Genet. 5, 22-29 (1993)
[ PubMed
].
14. Friedman LK, Koudinov AR.
Unilateral GluR2(B) Hippocampal Knockdown: A Novel Partial Seizure
Model in Young Rat. J Neurosci. 19, 9412-9425 (1999) [ PubMed
] [ Full
Text ] [ Reprint
Order ].
15. Koudinov AR, Koudinova
NV. Soluble amyloid beta protein is secreted by HepG2 cells as an apolipoprotein.
Cell
Biol Inter. 25, 265-271 (1997) [ PubMed
] [ Reprint
Order ].
16. Kaplan B, Haroutunian V,
Koudinov AR, Patael Y, Pras M, Gallo G. Biochemical assay for amyloid b
deposits to distinguish Alzheimer's disease from other dementias. Clin
Chim Acta 80, 147-159 (1999) [ PubMed
] [ Reprint
Order ].
17. Koudinov AR, Berezov TT,
Koudinova NV. The levels of soluble amyloid beta in different high density
lipoprotein subfractions distinguish Alzheimer's and normal aging cerebrospinal
fluid: implication for brain cholesterol pathology? Neurosci Lett. 314,
115-118 (2001) [ PubMed
] [ Full
Text ] [ PDF
] [ Reprint
Order ].
The above article presents experimental
data and discusses how an impairment of extracellular lipoprotein-mediated
trafficking of brain cholesterol may be linked to synaptic function, neural
apolipoproteins and Alzheimers disease
18. Koudinov AR, Koudinova
NV, Kumar A, Beavis R, Ghiso J. Biochemical characterization of Alzheimer's
soluble amyloid beta protein in human cerebrospinal fluid: association
with high density lipoproteins. Biochem Biophys Res Commun. 223,
592-597 (1996) [ PubMed
] [ Reprint
Order ].
19. ( 1 web+ citations )
Astrelin AV, Sokolov MV, Behnisch T, Reymann KG, Voronin LL. Principal
component analysis of minimal excitatory postsynaptic potentials. J
Neurosci Meth. 79, 169-186 (1998) [ PubMed
]; Samestkij EA, Bayazitov IT, Kleschnikov AM. AMPA and NMDA receptor
mediated components of "minimal" EPSPs recorded from the same synaptic
terminals show equal posttetanic LTP in the CA1 hippocampal region in vitro.
5th IBRO World Congress of Neuroscience, 11-15 July, 1999, Jerusalem, Israel.
Abstract book p. 191 (1999).
20. Ishida A, Furukawa K, Keller
J, Mattson MP. Secreted form of beta-amyloid precursor protein shifts the
frequency dependency for induction of LTD, and enhances LTP in hippocampal
slices. Neuroreport 8, 2133-2137 (1997) [ PubMed
].
21. Larson J, Lynch G, Games
D, Seubert P. Alterations in synaptic transmission and long-term potentiation
in hippocampal slices from young and aged PDAPP mice. Brain Res. 840,
23-35 (1999) [ PubMed
].
22. Xie Z, Sastry BR.
Induction of hippocampal long-term potentiation by alpha-tocopherol. Brain
Res. 604, 173-179 (1993) [ PubMed
].
23. ( 1 web+ citations ) Auerbach
JM, Segal M. Peroxide modulation of slow onset potentiation in rat
hippocampus. J Neurosci. 17, 8695-8701 (1997) [ PubMed
]; Cavus I, Teyler T. Two forms of long-term potentiation in area CA1 activate
different signal transduction cascades. J Neurophysiol. 76,
3038-3047 (1996) [ PubMed
].
24. Koudinov A, Groner Y, Segal
M. Cu/Zn-SOD transgenic mice are impaired in slow onset, long term potentiation.
Neurosci
Lett. 51, S23 (1998) [ Abstract
] [ Presentation
Order ].
25. Sato N, Kamino K, Tateishi
K, et. al. Elevated amyloid b protein (1-40)
level induces CREB phosphorylation at serine-133 via p44/42 MAP kinase
(Erk1/2)-dependent pathway in rat pheochromacytome PC12 cells. Biochem
Biophys Res Commun. 232, 637-642 (1997) [ PubMed
].
26. Segal M, Murphy DD. CREB
activation mediates plasticity in cultured hippocampal neurons. Neural
Plast. 6, 1-7 (1998) [ PubMed
].
27. Nitsch RM, Deng A, Wurtman
RJ, Growdon JH. Metabotropic glutamate receptor subtype mGluR1alpha stimulates
the secretion of the amyloid beta-protein precursor ectodomain. J Neurochem.
69,
704-712 (1997) [ PubMed
].
28. Cohen AS, Raymond CR, Abraham
WC. Priming of long-term potentiation induced by activation of metabotropic
glutamate receptors coupled to phospholipase. Hippocampus 8,
160-170 (1998) [ PubMed
].
29. Knowles RB, Wyart C, Buldyrev
SV, et al. Plaque-induced neurite abnormalities: implications for disruption
of neural networks in Alzheimer's disease. Proc Natl Aca Sci
USA 96, 5274-5279 (1999) [ PubMed
].
30. ( 4 web+ citations ) Selkoe
D. The origins of Alzheimer disease: a is for amyloid. JAMA.
283,
1615-1617 (2000) [ PubMed
] [ Full
Text ]; Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy.
Physiol
Rev. 81, 741-66 (2001) [ PubMed
]; Selkoe DJ. Toward a comprehensive theory for Alzheimer's disease. Hypothesis:
Alzheimer's disease is caused by the cerebral accumulation and cytotoxicity
of amyloid beta-protein. Ann N Y Acad Sci. 924, 17-25 (2000)
[ PubMed
]; Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's
disease Nature. Neurological disorders. 399 (Supplement),
A23-A31.
To moderate
the opinion expressed in cited above redundant articles ( Ref. 30 ) please
read contributions referenced below
31. ( 3 web+ citations ) Joseph J, Shukitt-Hale
B, Denisova NA, Martin A, Perry G, Smith MA. Copernicus revisited: amyloid
beta in Alzheimer's disease. Neurobiol Aging. 22, 131-146
(2001) [ PubMed
] [ Letter to the Editor ]; Mesulam MM. (1999) Neuroplasticity
failure in Alzheimer's disease: bridging the gap between plaques and tangles.
Neuron.
24,
521-529 (1999) [ PubMed
] [ Full
Text ]; Lue LF, Kuo YM, Roher AE, et al. Soluble amyloid beta peptide
concentration as a predictor of synaptic change in Alzheimer's disease.
Am
J Pathol. 155, 853-862 (1999) [ PubMed
].
RELATED
LETTERS TO EDITOR
[ Authors
"eLetters to Editor" collection ]
LE1. Koudinov AR, Koudinova
NV. Alzheimer's pathogenesis: tau and amyloid - a consensus or a challenge
for a third party quest ? Br Med J. E.letter published online September
4, 2001 [ Read
the letter ].
LE2. Koudinov AR, Koudinova
NV. Cholesterol, synaptic function and Alzheimer's disease. Br
Med J. E.letter published online October 16, 2001 [ Read
the letter ].
LE3. Koudinov AR, Koudinova
NV. Dementia, cholesterol and the soft science of dietary fat. Br
Med J. E.letter published online July 27, 2001 [ Read
the letter ].
LE4. Koudinov AR, Koudinova
NV. Looking forward for a historic issue, or: Is there anything besides
amyloid? Br Med J. E.letter published online October 19, 2001
[
Read
the letter ].
LE5. Koudinov AR, Koudinova
NV. Cholesterol supply and synaptic plasticity. Submitted to Science
on November 9, 2001 [ Read
the letter ].
LE6. Koudinov AR, Koudinova
NV. Cholesterol misregulation causes Alzheimer's disease: diet likely involved.
Br
Med J. E.letter published online December 1, 2001 [ Read
the letter ].
SUPPLEMENTARY
REFERENCES
1S. Jang HF. Measurement
of Fibril Angle in Wood Fibres with Polarization Confocal Microscopy. J
Pulp Paper Science 24, 224-30 (1998).
2S. Ferris SH, Kluger
A. Assessing cognition in Alzheimer disease research. Alzheimer Dis
Assoc Disord. 11S, 45-49 (1997) [ PubMed
].
3S. Franssen EH, Reisberg
B. Neurologic markers of the progression of Alzheimer's disease. Int
Psychogeriatr. 9 (Suppl 1), 297-306; discussion 317-21 (1997)
[ PubMed
].
4S. Chen G, Chen KS, Knox
J, et al. A learning deficit related to age and beta-amyloid plaques in
a mouse model of Alzheimer's disease. Nature 408, 975-979
(2000) [ PubMed
].
5S. Koudinov AR, Berezov
TT. Alzheimer's amyloid plaque formation is a condition for neuronal dysfunction.
10th European Neurological Society Meeting, 18 June, 2000, Jerusalem, Israel.
Abstract book. P534 (2000) [ Abstract
].
6S. Koudinov AR, Koudinova,
NV, Berezov TT. Is Alzheimer's amyloid plaque formation a condition for
neuronal dysfunction? Neurobiol. Aging 21, S155 (2000) [ Abstract
].
 CLICK
HERE OR PRESS <CTRL><D> TO BOOKMARK THIS ARTICLE FOR FUTURE REFERENCES CLICK
HERE OR PRESS <CTRL><D> TO BOOKMARK THIS ARTICLE FOR FUTURE REFERENCES |
Readers statistics:
|
Citation example for this article:
Koudinov AR, Berezov TT, Koudinova NV. Amyloid plaque (and not diffuse
amyloid) is a condition for neuronal dysfunction. Clin. Med. Health
Res. published online December, 2001, clinmed/2001110002, http://clinmed.netprints.org/cgi/content/full/2001110002v1
You
may also wish to cite year 2000 preliminary accounts of this article
[ 5S or 6S ].
HTML/PDF
article style design & programming: © 2001 Alexei Koudinov ||
Want to make your ClinMed Netprint
article looking like ours? Call
for advise/templates
